Home
Tutorial: Rigid Body Refinement
Contributors: Kristian Lytje, Jan Skov Pedersen, Andreas Haahr Larsen, Jeppe Breum Jacobsen.
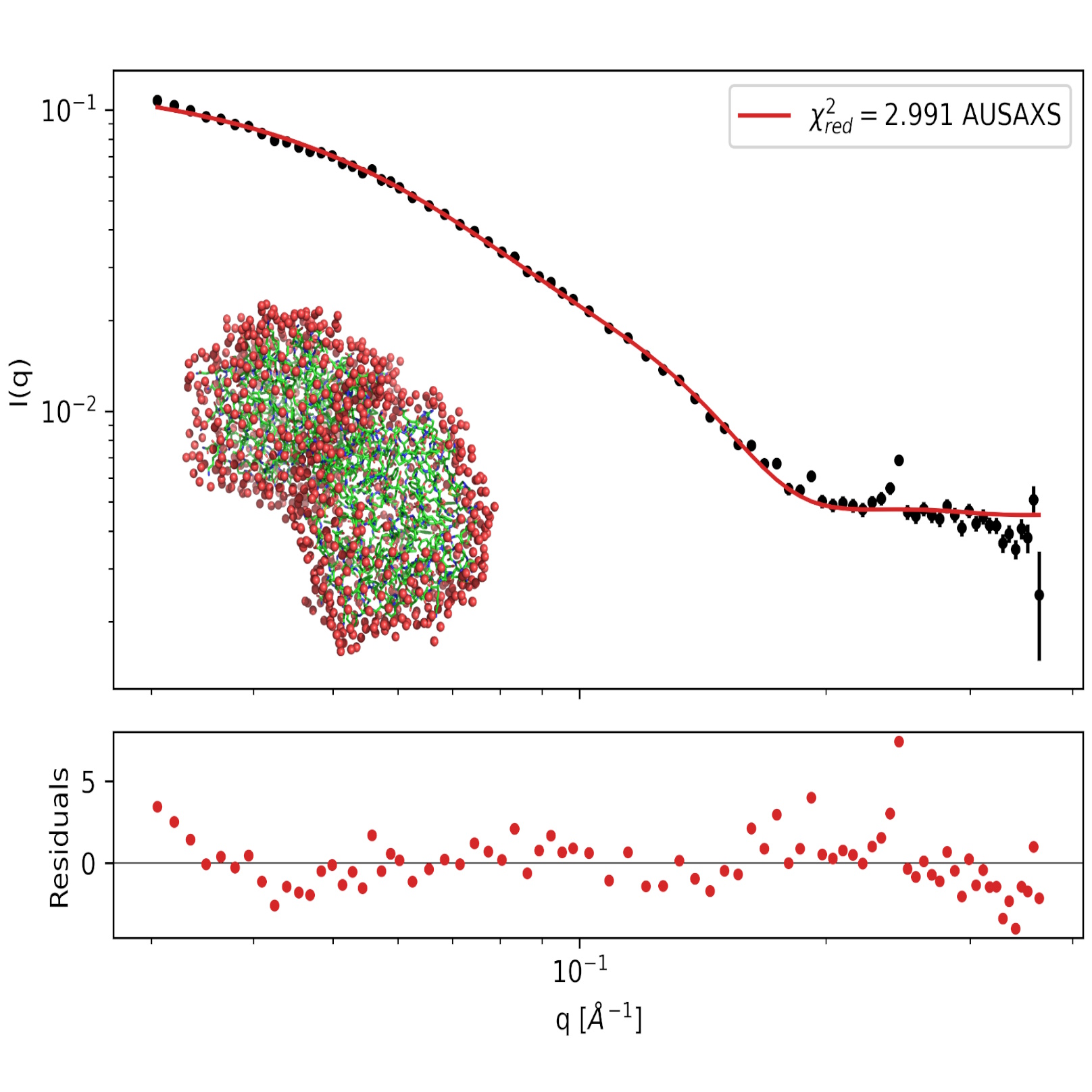
SAXS data on the PA2 protein in solution with a fitted hydration layer.
Before you start
- For an introduction to analysis of SAXS data on proteins in solution, we recommend the proteins tutorial.
- This tutorial uses the
rigidbody_optimizerfrom AUSAXS.
Other programs, such as SASREF, which is part of the ATSAS package, can also perform rigid body refinements. - To visualize molecular structures, a program such as PyMOL can be used, which has a free 30-day trial.
Alternatively, the RCSB protein data bank has an online 3D viewer.
Learning outcomes
After completing this tutorial you will:- Know what rigid body modeling is.
- Be able to use SAXS data to optimize a rigid body model with AUSAXS.
- Be able to visualize your structures with PyMOL.
- Optionally, be able to use AlphaFold to generate a structural hypothesis.
Introductory remarks
Pea albumin (PA) is a protein found in peas, and comes in two forms, PA1 and PA2. In solution, PA2 aggregates to form a dimer. In this tutorial, which is based on the paper by Ruifen Li et al, 2025, we will look at how SAXS data can be used to optimize a rigid body model of PA2.
Part 0: Installing AUSAXS
We will in this tutorial use AUSAXS to analyze our SAXS data, and later to perform the rigid body refinement.
It may be installed in two ways:
- Using Python:
pip install pyausaxs. This will install the command-line toolausaxs. -
Manual download: Download the latest version from GitHub and unzip it in a desired location.
While this will also give you access to the program, you will need to manually provide the explicit path to the unzipped location when running the program from the command line. For example, if you unzipped it inC:\AUSAXS, you would runC:\AUSAXS\rigidbody_optimizer.exeto run the rigid body optimizer.
Part I (OPTIONAL): Use AlphaFold to generate hypothetical structure
AlphaFold is a machine learning algorithm that can, among other things, predict the structure of a protein based on its amino acid sequence. It can be run by anyone with a Google account on the AlphaFold Server. Later in this tutorial, we will use these structures for fitting our SAXS data. If you are not interested in using AlphaFold, you can skip to the next part of the tutorial, where the structures will be provided.
-
Generate protein structures with AlphaFold
- Before using AlphaFold, we need the protein sequence in FASTA format. Luckily PA1 and PA2 have already been sequenced. - Go to the UniProt entry for PA1 and find the sequence. The sequence for PA1 contains a signal peptide, which is not present in the mature protein. Remove the signal peptide by disregarding the first 26 amino acids.
- With the sequences of PA1 and PA2, go to the AlphaFold Server and login with a Google account.
- For PA1, paste the sequence in the input box, and click "Continue and preview job". Remember to not include the signal peptide!
- For PA2, paste the sequence in the input box and click "Continue and preview job". (The dimer-structure is added by the rigid body modeling later.)
- Once AlphaFold has finished, the folded proteins can be viewed, and you can download a zip-file. The zipfile contains a number of files, but we are interested in the .cif files, which contain the 3D coordinates of the protein structure. Give the structures meaningful names.
- Go to the UniProt entry for PA2 and find the sequence.
If the AlphaFold preview of the structure does not work, you can open the .cif files in PyMOL to visualize the structures.
Part II (Optional): PA1 warmup
To familiarize ourselves with the AUSAXS package, we will first look at the PA1 protein, which is a monomer. If you have already completed the proteins tutorial and used the SAXS fitter from AUSAXS, you can skip this part. The goal now is to use the SAXS fitter from AUSAXS to test the agreement between the SAXS data and the AlphaFold structure of PA1. The program calculates the scattering from the provided structure and fits a hydration layer. The program provides a graph showing the fit and the data as well as the reduced chi square ($\chi^2_{red}$).
-
Fit a SAXS dataset to a protein structure
- Download the SAXS data file for PA1. If you did not generate a structure for PA1 with AlphaFold, you can download the .cif file here.
- Open a command window (Terminal on MACOS). Type
ausaxs fit PA1_no_signal_peptide.cif PA1.datand press Enter. - The program will now perform the fitting procedure. When it is done, you should find find a folder
output\saxs_fitter\PA1, with a number of graph instruction files. To convert these into actual graphics, runausaxs plot. - Among other things, the plotting-program provides the usual log- and loglog-plots, which show the $\chi^2_{red}$. The program also provides a a
model.pdbfile, which can be opened in PyMOL. Opening this alongsidePA1_no_signal_peptide.cifallows you to see the hydration layer added by the fitting procedure.
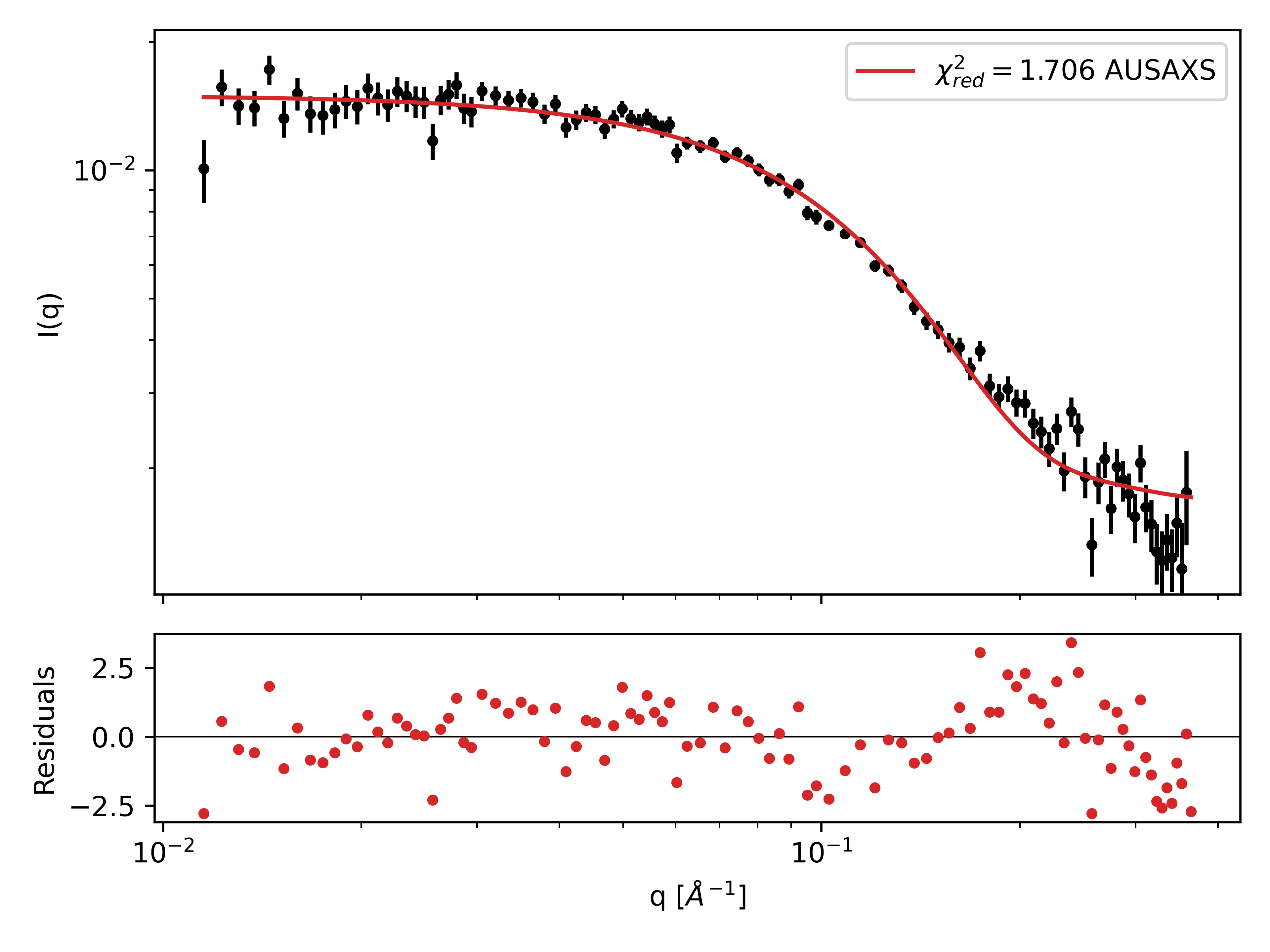
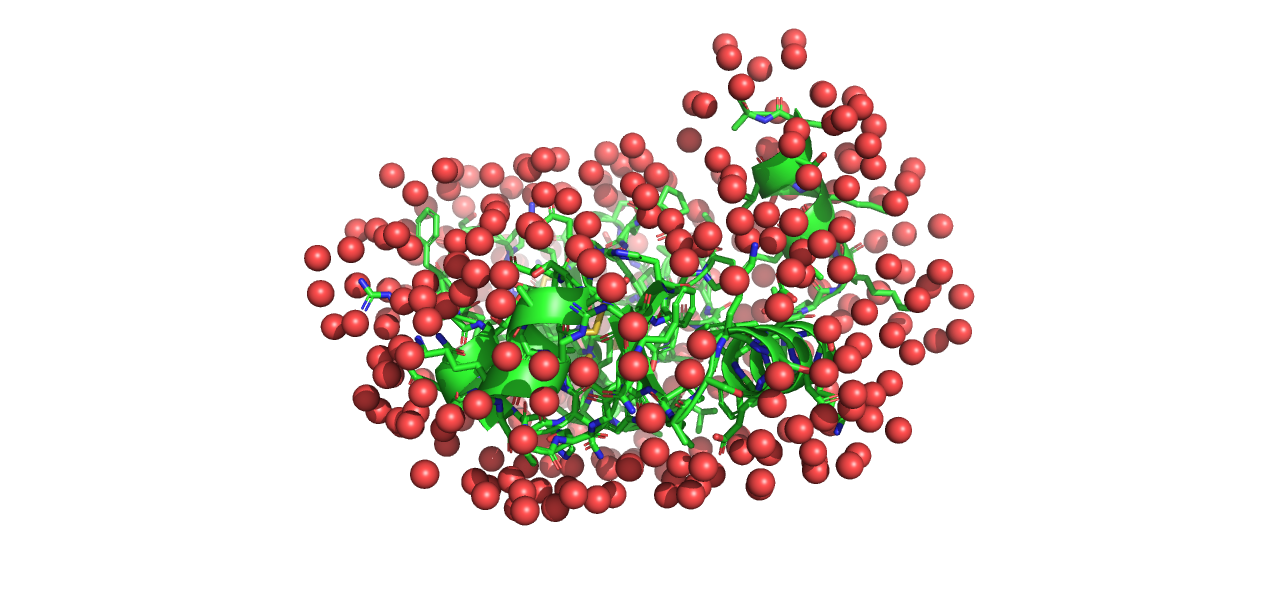
Part III: Rigid body refinement of PA2
In this part, we will look at the PA2 dimer. PA2 is known to form dimers in solution. It is possible for AlphaFold to generate a dimer structure, but instead we will use the monomer structure from AlphaFold and identify the dimer structure through rigid body refinement against the SAXS data. Rigid body refinement is a method where two or more subunits are treated as rigid bodies, and their relative positions and orientations are optimized while leaving the internal structure intact.
-
Fit a SAXS dataset to a protein structure
- Download the SAXS data file for PA2. If you did not generate the folded dimer structure for PA2 in part I, you can download the .cif file here.
- To run the optimizer, we need to create a configuration file that tells the program what to do. We will later create one ourselves, but for now, you can download this config file and place it in the same directory as the data files.
- Open the
config.conffile with a text editor and edit the paths to your SAXS data file and structure file. - Before saving and closing the config file, look at its contents. It tells the optimizer to create a dimer structure with P2 symmetry, and try to optimize it 500 times. For more information, see the wiki page on GitHub.
- To run the program, open a command window and navigate to the directory with the data. Type
ausaxs rigidbody config.conf. In the command window, you can confirm that the data and structure are loaded correctly. Each time the program finds a new dimer structure that improves the $\chi^2$ value, it prints it in the command window. - When the program is finished, it prints a fit report that tells you the final $\chi^2$ value and other relevant information.
- In the directory where you ran the program, you should find the unoptimized dimer structure
initial_state0.pdband the optimized structurefinal_state1.pdb. To see how your final structure compares to the SAXS data, runausaxs plot, and find the log.png and loglog.png graphs in the output folder. - Finally, open PyMOL and load
initial_state0.pdbandfinal_state1.pdbto visualize the change made by the program.

Exercise
- Earlier we mentioned that AlphaFold can also generate dimer structures. So the question is, how do these predicted dimer structures compare to the ones obtained through rigid body refinement? To test this, generate a structure for PA2 with AlphaFold as in part I, but with "copies" set to 2. Run this structure along with the SAXS data in the SAXS fitter. Compare the log.png or loglog.png graph to the ones you got from the rigid body refinement. Also check the $\chi^2$ values.
- You can also directly refine the dimer structure. Modify the script to use the dimer structure from AlphaFold as the initial structure, and run the rigid body optimizer. Does it improve the fit? How does the final structure compare to the one obtained from the monomer structure? With AUSAXS, symmetries are generally preferred for efficiency.
Challenges
As another example of what can be done with rigid body refinement, refine the structure of the flexible human leucocyte common antigen-related protein.
Download the files LAR.pdb and LAR.dat, and use the rigid body optimizer to refine the structure.
To get started, inspect the structure in PyMOL. Mark (click) all points in the structure you think look flexible; these will later be used to define the rigid bodies.
To obtain the sequence numbers of these points, open the "Display" menu and select "Sequence". This will show the sequence of the protein, with your previous selections highlighted.
With this, you have all the information you need. Define your configuration file, and run the optimizer.
Hints:
- You will need to use the "split" option in the
load element. - Remember to define constraints between each pair of bodies. For this, the
autoconstrain linearmay prove useful. - You will need a few thousand iterations in total to properly converge. You can also experiment with mixed transformation modes.
- The article mentions discarding data points smaller than $q=0.0125$ due to aggregation issues. You should follow this suggestion for best results.
They also mention that the final structure has an "L"-shape. Does your final structure have the same shape?
Perspectives
For more information on the interplay between AlphaFold and SAXS data, see the tutorial on the SAXSAFOLD website.
Help and feedback
Help us improve the tutorials by- Reporting issues and bugs via our GitHub page. This could be typos, dead links etc., but also insufficient information or unclear instructions.
- Suggesting new tutorials/additions/improvements in the SAStutorials forum.
- Posting or answering questions in the SAStutorials forum.